Part 1 of 5-Part Series: Design Inputs - The Foundation for Effective Medical Device Development

rmforge.io | Part 1 of a 5-Part Series on Design Controls
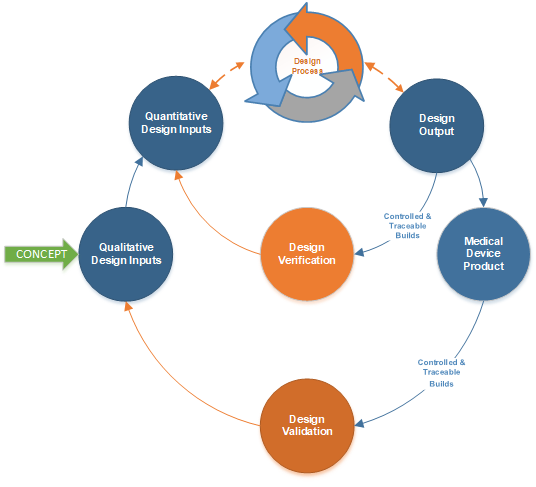
After conceiving a new medical device design, the next move for efficient product development is to initiate design controls through documented Design Inputs. Design Inputs are the written translation of user needs and intended uses into product requirements. Documented, released into a controlled and traceable system, they tell the team exactly what is being designed.
Projects run faster and cost less when Design Inputs are documented from the earliest possible stage. Specific goals, clear communication, fewer mistakes. The objection to early documentation is usually some version of "things will change." That objection is wrong. Change is the reason for early documentation, not the argument against it. Controlled changes with focused reviews and on-the-spot approvals move faster than uncontrolled drift.
The biggest misstep in early project stages is delaying Design Input documentation and using inconsistent naming conventions. Both create design target ambiguity ("Where is the target?") and prolong timelines from R&D through regulatory submissions. It happens so regularly in early-stage projects that it has become more the norm than not.
A Familiar Story
After a team meeting, an engineer designs and prototypes a vascular stent for 400 million cycles based on the discussion and a push to complete testing in under six months. The project manager actually wants 600 million cycles based on executive feedback and company strategy but never documents it, assuming the team picked it up from prior meetings. The team spends money making stents and starting durability testing prematurely against undocumented requirements. The Vice President finds out the stent failed between 300 million and 350 million cycles, three and a half months in. Problems on multiple levels, not least: why were Design Inputs never documented, and was the durability equipment ever test method validated?
Early documentation of Design Inputs with standardized naming, ideally drawn from regulations and standards, gives the team clarity, regulatory alignment, and a common language. That sets design expectations and minimizes redo cycles and delays.
Two Categories. Just Two.
Design Inputs split into two primary groups:
- Qualitative Inputs: User Needs and Intended Uses
- Quantitative Inputs: Product Requirements
Do not overthink it. There are only two kinds. Why? Downstream there are only two kinds of design testing: Design Verification and Design Validation (DV&V). The Systems Engineering folks will lobby for System Requirements as a third category. Keep it to two categories and the project moves smoothly even on complex systems. DV&V testing will also make more sense.
Common Alternate Names (Watch For These)
User Needs and Intended Uses are sometimes called Clinical Requirements, Clinical Specifications, User Requirements, Marketing Requirements, or Marketing Specifications. Standards basis: FDA 21 CFR 820.10(c) and ISO 13485:2016 Section 7.3.3. (Note: under the QMSR effective February 2, 2026, the legacy 820.30 subsections are [Reserved].)
Product Requirements are sometimes called Physical and Performance Requirements, Technical Requirements, Engineering Specifications, System Requirements, Functional Requirements, or Product Specifications. Standards basis: same as above. EU MDR 2017/745 does not explicitly label "Design Input" and "Design Output" but requires equivalent evidence within General Safety and Performance Requirements (Annex I) and Technical Documentation (Annex II).
Note on terminology: Product Specification is an output-side label. Within design inputs, the best-practice label is Product Requirement. The phase context matters. (See the separate rmForge blog: Product Requirement vs Product Specification.)
Example Design Inputs (Vascular Stent)
Example only. Not representative of any real product.
ID | User Need and Intended Use | Product Requirement | Standards |
UN1 / PR1 | The stent shall be biologically safe. | The stent shall pass biocompatibility, chemical characterization, and extractable and leachable testing per ISO 10993-1 Table A.1: Implant, Blood Contact, Permanent (>30d). | ISO 10993-1 |
UN2 / PR2 | The stent shall be sterile. | Sterility Assurance Level (SAL) shall be 1x10-6 CFU. | ISO 11135 |
UN3 / PR3 | Device life in years shall meet standardized requirements and regulatory guidance. | Simulating 10 years equivalent device life, the stent shall survive 600 million cycles minimum of durability testing through 2-5% radial displacement and 2.00 +/- 0.25 mm axial displacement at 40 Hz maximum. | ISO 25539, ASTM F2942, ASTM F2477 |
UN4 / PR4 | The stent shall be transcatheter-delivered by an interventional cardiologist. | Guide catheter OD shall be 6 Fr maximum. Catheter length shall be 100 +/- 1 cm. | ISO 10555-1 |
Writing User Needs and Intended Uses
These are typically qualitative, describing what the user needs to achieve and how the device is intended to be used. Sources include physician input, marketing, clinical literature, post-market feedback, and competitor analysis from publicly available sources. They usually do not have explicit quantification, but there is no prohibition against including it.
Keep them high-level. The validation test is whether they can be Design Validated through simulated use, preclinical study, or clinical trial. If a User Need cannot be validated, rewrite it.
Writing Product Requirements
Product Requirements are quantitative. They specify measurable design targets, tolerances, and performance parameters. They are tangible translations of User Needs into bench-testable requirements.
Two filters before publishing any Product Requirement:
- Is it testable? Can the team measure it? Not theoretically. With the test setup actually available to the team.
- Is the test setup capable of measuring it? Continuing the stent example: a Product Requirement of 2.00 +/- 0.05 mm displacement requires a test system that can measure displacement at a precision well inside that range. A wooden ruler would consume the entire span of the requirement with measurement error. A laser micrometer or vision-based system is more appropriate. This is the test method validation (TMV) question.
Core Considerations
Specificity and Standardization
Each requirement should be succinct and detailed enough to enable verification and validation testing. Use standard terminology from FDA, ISO, IEC, AAMI, and ASTM wherever possible. Avoid coining new vocabulary.
Traceability
Every requirement gets a unique identifier for traceability through later stages. The Design Input and Design Output Matrix (Part 5) is where this lives.
Regulatory Alignment
Align documentation with the QMSR (21 CFR 820.10(c)) and ISO 13485:2016 Section 7.3.3. Lack of clarity equals confusion, which equals questions from regulators and auditors.
Change Management
Every update to a Design Input gets reviewed and approved per 21 CFR 820.10(c) and ISO 13485:2016 Section 7.3.9. Do not be reluctant to make changes based on new evidence. That is part of the Design Control process.
The Bottom Line
Categorizing into Qualitative Design Inputs and Quantitative Design Inputs creates a clear lineage from high-level expectations to measurable criteria. That lineage flows into Design Outputs (Part 2), gets tested through DV&V (Part 3), and connects to risk management (Part 4). Documenting Design Inputs, standardizing naming, and aligning with regulations sets up a streamlined and compliant product development process. Lower risk. Lower cost. Better timelines.
The 5-Part Series
- Part 1: Design Inputs (this article)
- Part 2: Design Outputs
- Part 3: Design Verification and Design Validation
- Part 4: Risk Management in Design Control
- Part 5: Design Input and Design Output Matrix
References
Quality Management System Regulation (QMSR), 21 CFR Part 820, Feb. 2, 2026.
ISO 13485:2016. Medical Devices, Quality Management Systems, Requirements for Regulatory Purposes. ISO, 2016.
ISO 14971:2019. Medical Devices, Application of Risk Management to Medical Devices. ISO, 2019.
Design Control Guidance for Medical Device Manufacturers. FDA, 1997.
Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on Medical Devices. Official Journal of the European Union, 2017.
ISO 10993-1:2018. Biological Evaluation of Medical Devices, Part 1: Evaluation and Testing Within a Risk Management Process.
ISO 11135:2014. Sterilization of Health Care Products, Ethylene Oxide.
ISO 25539 series. Cardiovascular Implants, Endovascular Devices.
ASTM F2942-19. Standard Guide for In Vitro Axial, Bending, and Torsional Durability Testing of Vascular Stents.
ASTM F2477-19. Standard Test Methods for In Vitro Pulsatile Durability Testing of Vascular Stents.
ISO 10555-1:2023. Intravascular Catheters, Sterile and Single-Use Catheters, Part 1: General Requirements.
FDA Guidance. Non-Clinical Engineering Tests and Recommended Labeling for Intravascular Stents and Associated Delivery Systems. FDA, April 18, 2010.
Questions or feedback: mike@rmforge.io
Try the platform: rmforge.io